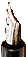

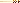
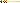
Among human beings, sickle-cell anemia is a particularly well-studied case of adaptation (Figure 2.7). This painful disease, in which the oxygen-carrying red blood cells
Normal Allele
Codon Amino acid Position
Codon
Figure 2.7 Mutation of a single base of DNA can result in a dramatically different protein. Pictured here are codons 3 through 9 for the beta chain of hemoglobin, the protein that carries oxygen in red blood cells and the amino acids these codons specify. The top row depicts the normal allele, and in the bottom row is the single substitution that makes the red blood cells bend into a sickle shape (clogging the capillary beds and causing great pain, which is what occurs with sickle-cell anemia). Sickling occurs because the amino acid valine, compared to glutamic acid in the normal allele, gives the hemoglobin molecule different properties. The beta chain is 146 amino acids long. A simple mutation (the substitution of thymine for adenine in position 6 as indicated in red) has dramatic and tragic consequences.

Sickle-cell anemia is caused by a genetic mutation in a single base of the hemoglobin gene resulting in abnormal hemoglobin, called hemoglobin S. Those afflicted by the disease are homozygous for the S allele, and all their red blood cells “sickle.” Co-dominance is observable with the sickle and normal alleles. Heterozygotes make 50 percent normal hemoglobin and 50 percent sickle hemoglobin. Shown here is a sickle hemoglobin red blood cell among normal red blood cells.
Change shape (sickle) and clog the finest parts of the circulatory system, is caused by a mutation in the gene coding for hemoglobin, the protein responsible for oxygen transport. This disorder first came to the attention of geneticists in Chicago when it was observed that most North Americans who suffer from it are of African ancestry. Investigation traced the abnormality to populations that live in a clearly defined belt across tropical Central Africa where the sickle-cell allele is found at surprisingly high frequencies. Geneticists were curious about why such a harmful hereditary disability persisted in these populations.
According to the theory of natural selection, any alleles that are harmful will tend to disappear from the group, because the individuals who are homozygous for the abnormality generally die—are “selected out”—before they are able to reproduce. Why, then, had this seemingly harmful condition remained in populations from tropical Central Africa?
The answer to this mystery began to emerge when it was noticed that the areas with high rates of sickle-cell anemia are also areas in which a particularly deadly form of malaria (falciparum) is common (Figure 2.8). This severe form of malaria causes many deaths or, in those who survive, high fever that significantly interferes with the victims’ reproductive abilities. Moreover, it was discovered that hemoglobin abnormalities are also found in people living in parts of the Arabian Peninsula, Greece, Algeria, Syria, and India, all regions where malaria is (or was) common.
Increased ability to survive the effects of the malarial parasite; it seems that the effects of the abnormal hemoglobin in limited amounts were less injurious than the effects of the malarial parasite. Thus selection favored heterozygous individuals with normal and sickling hemoglobin (HbAHbS). The loss of alleles for abnormal hemoglobin
Further research established that while individuals with hemoglobin abnormalities can still contract malaria, hemoglobin abnormalities are associated with an
Sickle-cell anemia An inherited form of anemia caused by a mutation in the hemoglobin protein that causes the red blood cells to assume a sickle shape.

Figure 2.8 The allele that, in homozygotes, causes sickle-cell anemia makes heterozygotes resistant to falciparum malaria. While falciparum malaria is also found is tropical Latin America, the sickle cell allele is most common in populations native to regions of the Old World where this strain of malaria originated.
Caused by the death of those homozygous for it (from sickle-cell anemia) was balanced out by the loss of alleles for normal hemoglobin, as those homozygous for normal hemoglobin were more likely to die from malaria and to experience reproductive failure.
Expression of normal versus sickle hemoglobin in a heterozygous individual represents an example of incomplete dominance. The mutation that causes hemoglobin to sickle consists of a change in a single base of DNA, so it can arise readily by chance (see Figure 2.7). The resulting mutant allele codes for an amino acid substitution in the beta chain of the hemoglobin protein that leads red blood cells to take on a characteristic sickle shape. In homozygous individuals with two sickle-hemoglobin alleles, collapse and clumping of the abnormal red cells block the capillaries and create tissue damage—causing the symptoms of sickle-cell disease. Afflicted individuals commonly die before reaching adulthood.
The homozygous dominant condition (HbAHbA— normal hemoglobin is known as hemoglobin A, not to be confused with blood type A) produces only normal molecules of hemoglobin whereas the heterozygous condition (HbAHbS) produces some percentage of normal and some percentage of abnormal hemoglobin. Except under low oxygen or other stressful conditions, such individuals suffer no ill effects. The heterozygous condition can actually improve individuals’ resilience to malaria relative to the “normal” homozygous condition.
This example also points out that adaptation tends to be specific; the abnormal hemoglobin was an adaptation to the environment in which the malarial parasite flourished. When individuals who had adapted to malarial regions came to regions relatively free of malaria, what had been an adaptive characteristic became an injurious one. In environments without malaria, the abnormal hemoglobin becomes comparatively disadvantageous. Although the rates of the sickle-cell trait are still relatively high among African Americans—about 9 percent show the sickling trait—this represents a significant decline from the approximately 22 percent who are estimated to have shown the trait when African captives were shipped across the Atlantic and sold as slaves. A further decline over the next several generations is to be expected, as selection relaxes for the frequency of the sickle-cell allele.
This example also illustrates the important role culture may play even with respect to biological adaptation. In Africa, the severe form of malaria was not a significant problem until humans abandoned food foraging for farming a few thousand years ago. In order to farm, people had to clear areas of the natural forest cover. In the forest, decaying vegetation on the forest floor made the ground absorbent, so the heavy rain rapidly soaked into the soil. But once stripped of its natural vegetation, the soil lost this quality. Also, without the forest canopy to break the force of the rainfall, strong rains compacted the soil further. As a result, stagnant puddles commonly formed after downpours, providing the perfect breeding environment for the type of mosquito that hosts the malarial parasite. These mosquitoes began to flourish and transmit the malarial parasite to humans. Thus humans unwittingly created the environment that made a previously disadvantageous trait, the abnormal hemoglobin associated with sickle-cell anemia, advantageous. While the biological process of evolution accounts for the frequency of the sickle-cell allele, cultural processes shape the environment to which humans adapt.
 World History
World History









